
Optimized prediction of cardiac cis-regulatory elements
We are building a catalogue of regulatory elements in human heart development at single-cell resolution, combining multi-omics data analysis and machine-learning approaches.
We are also implementing models to predict the impact of genetic variation on the activity of these elements. The project is part of the HeartX project (funded by the Swiss National Science Foundation), a collaboration with the labs of Marco Osterwalder (https://www.osterwalderlab.com/) and Christian Zuppinger at the Department for BioMedical Research (DBMR), University of Bern.
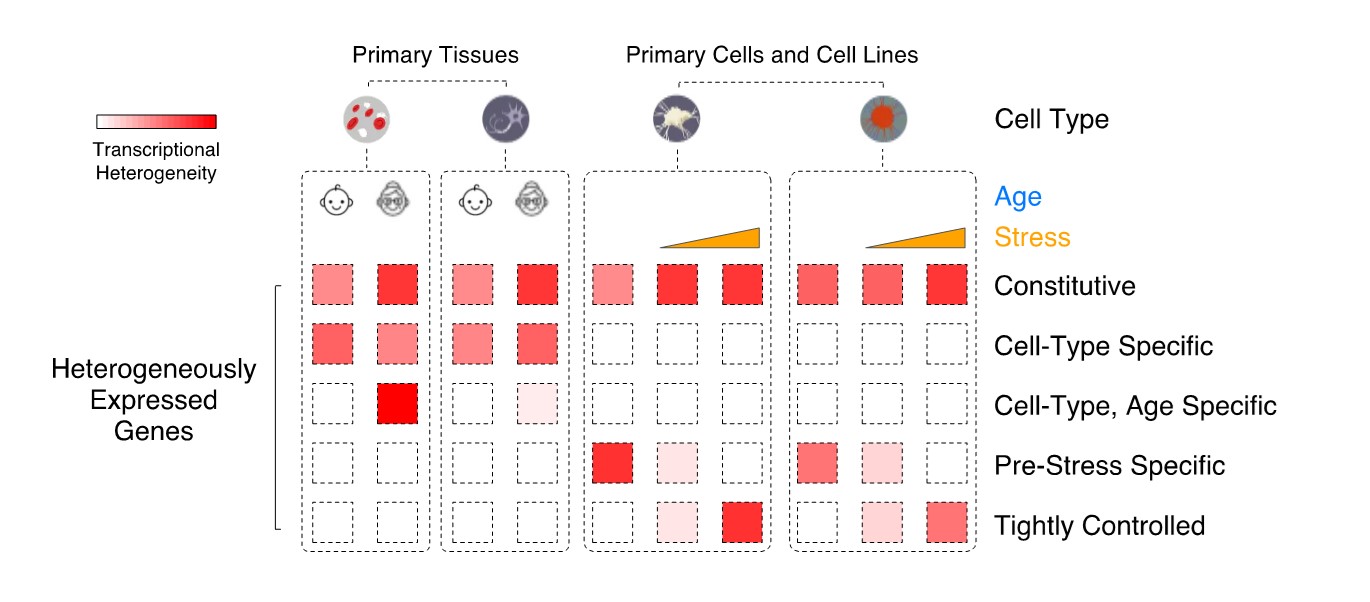
Regulated transcriptional noise
Leveraging single-cell transcriptional data, we aim at classifying mammalian genes based on their pattern of transcriptional heterogeneity across cells of the same type, in both normal tissues and disease states (Figure 1), with the ultimate goal of identifying if these differences in variability are driven by distinct modes of regulation.
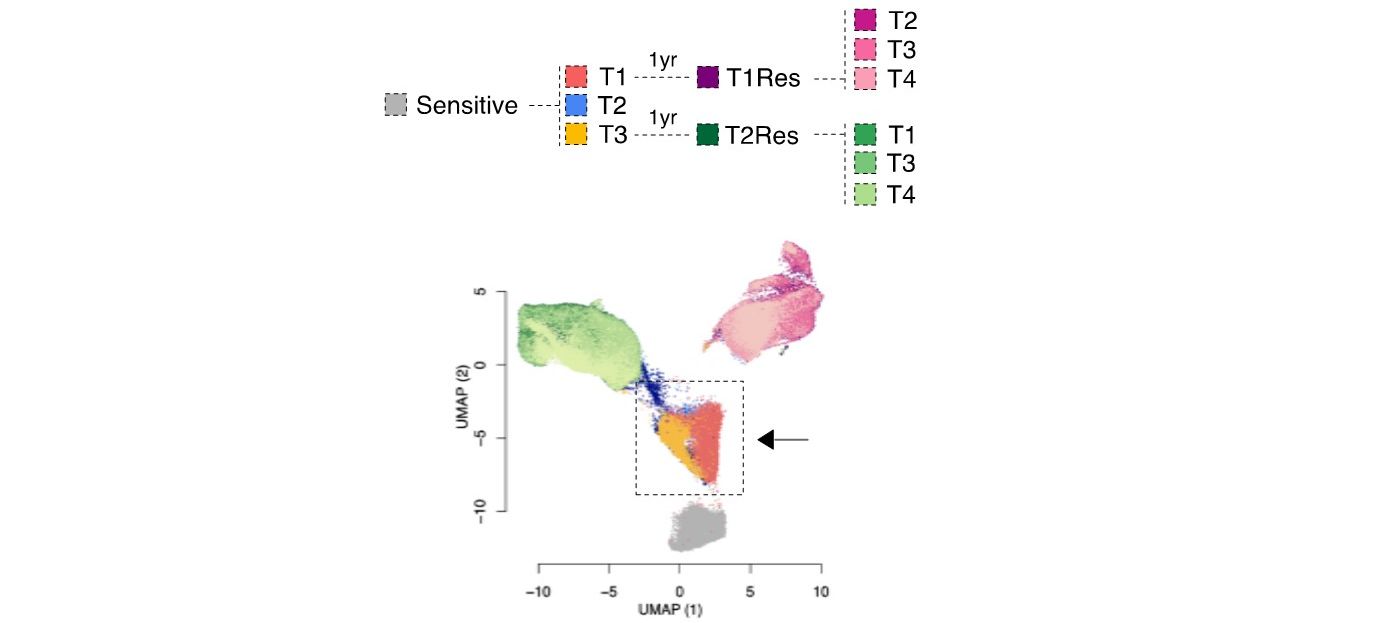
A conserved survival mechanism of drug-tolerant persister cells
We previously identified a transcriptional signature that predicts the phenotype of those luminal breast cancer cells surviving short-term endocrine therapies (Hong et al. 2019). These cells, often called drug-tolerant persisters, will eventually lead to relapse.
We aim at:
- investigating the level of conservation of this signature across tumors of different origin and its importance in survival to different therapeutic strategies, and
- characterizing its upstream regulators.
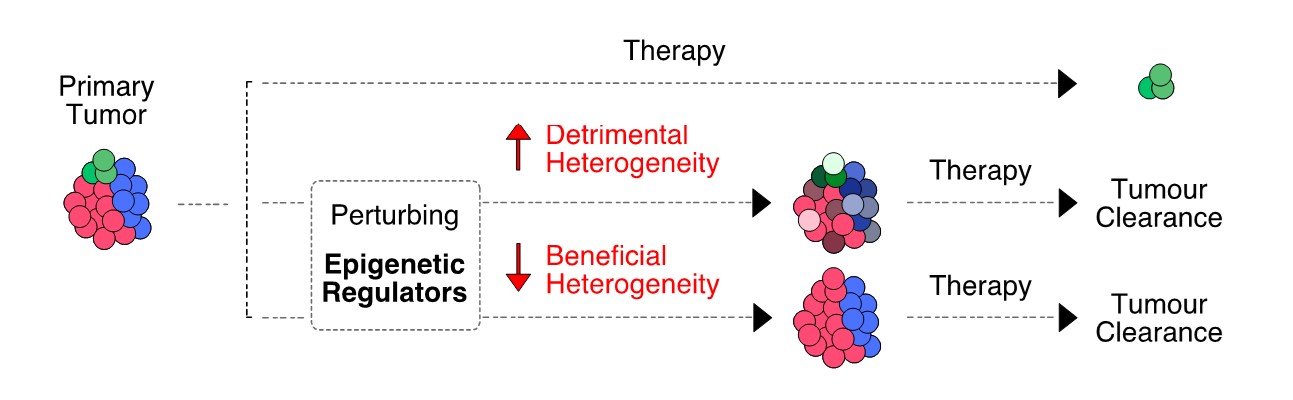
Manipulating phenotypic heterogeneity to delay tumor progression
The genes of the so-called epigenetic regulators are often mutated or mis-regulated in cancer. We hypothesize that their aberrant activity is associated to transcriptional fluctuations that could be manipulated to the patient’s advantage (Figure 3). Based on this rationale, we are combining in vitro perturbations (directed at epigenetic regulators) with single-cell transcriptomics, to pinpoint novel treatment strategies to delay tumor progression.
Development of new methods to identify the upstream regulators of gene activity
We are leveraging previously published, validated regulatory interactions to train machine learning models to then predict novel, tissue-specific interactions genome-wide. To this end, we are combining co-expression networks, transcription factor binding profiles and local, chromatin conformation measurements.